|
|
| chimica |
|
|
|||||
|
Metodi di deuterazione
1.2 Introduzione
La deuterazione dei composti aromatici
2.2.1 Reazione di scambio H-D sui derivati della piridina
2.2.2 Reazione di scambio H-D di indoli derivati
2.2.3 Reazione di scambio H-D dei derivati della Pirimidina ed immidazolo
2.2.4 Reazione di scambio H-D di composti biologicamente attivi
Scambio di deuterio acido-catalizzato che coinvolgono ioni di carbonio
Deuterazione base catalizzata di chetoni metilici
Riferimenti bibliografici
Da tempo, la conoscenza del rapporto isotopico D/H nei vari siti molecolari di una data specie viene usata per fornire una base di informazioni per lo studio dell'origine territoriale, della specie varietale e delle manipolazioni operate dall'uomo nel corso della lavorazione e conservazione del prodotto.
Fino a pochi anni fa, l'analisi dell'impronta isotopica del deuterio è stata utilizzata per l'analisi dei vini o di altri componenti molecolari contenuti in prodotti alimentari di origine vegetale e ha permesso di ottenere informazioni utili non solo per l'identificazione del sito territoriale di origine ma anche per la valutazione della qualità dei prodotti alimentari, del loro anno di produzione o di eventuali sofisticazioni ottenute per aggiunta di componenti di differente origine naturale o sintetica.
La metodologia di indagine SNIF-NMR è stata affinata e utilizzata per lo studio delle miscele di trigliceridi dell'olio di oliva extravergine, di oli vegetali di varia natura, e per l'analisi dello squalene contenuto nell'olio di oliva e di diversa origine[59, 60].
Diversi composti organici deuterati contenenti almeno il 99% di atomi di deuterio sono stati necessari per le indagini attualmente in corso da Steacie ed i suoi collaboratori[61].
I composti contrassegnati con deuterio hanno trovato ampie applicazioni in aree di ricerca come quella farmaceutica, bioanalitica, biologica, chimica e ambientale per analizzare i meccanismi del metabolismo dei farmaci, dei residui chimici negli alimenti usando la spettrometria di massa (MS), la struttura di biomolecole, e così via[62,63].
L'accesso dei composti contrassegnati con deuterio è stato facilitato con l'adozione del post-sintetico scambio di reazione H-D al posto del laborioso e costoso processo sintetico multi-step a partire originariamente dal deuterio.
Di recente è stato messo a punto una reazione di scambio H-D regioselettiva in posizione benzilica utilizzando Pd /C, un catalizzatore in ossido di deuterio sotto atmosfera di idrogeno (Pd/C-H2-D2O sistema) a temperatura ambiente[64], e si è riscontrato che l'applicazione di calore potrebbe promuovere lo scambio di reazione H-D non solo in posizione benzilica, ma anche sul carbonio non attivato[65]. La deuterazione eterociclica è molto importante soprattutto in composti come indolo, piridina, pirimidina, chinolina ed i sistemi di anelli eterociclici che sono presenti in prodotti naturali, prodotti farmaceutici, medicinali veterinari, agrochimici e così via.
Prendendo in considerazione l'istituzione di un metodo generale di deuterazione utilizzando il sistema Pd/C-H -D2O, si è studiato la deuterazione di una vasta gamma di substrati eterociclici.
Lo
scambio dell'atomo di idrogeno di composti aromatici con D2O è stato
migliorato utilizzando la catalisi di un supporto polimerico di acido solfonico
(Deloxan). Fenolo, anilina, chinolina e idrocarburi aromatici sostituiti sono
stati selettivamente perdeuterati sull'anello in alta resa, con significativa
formazione di prodotto a

. 2.1 Struttura dell'etilbenzene deuterato
Lo scambio H-D acido-catalizzato di substrati aromatici ed eteroaromatici è stato studiato fin dal 1960. I più comuni catalizzatori impiegati sono stati: acidi minerali concentrati e acidi di Lewis altamente reattivi, come gli alogenuri di organoalluminio o gli acidi carbossilici fluorinati. In ogni caso, tutti questi reagenti presentano degli inconvenienti e lo scambio H-D resta ancora un attivo campo di ricerca.
L'attuale ricerca sullo scambio H-D/T nei composti aromatici coinvolge l'utilizzo di catalizzatori acido solfonico polimero D2O, oppure richiede condizioni di reazione supercritiche. Ad elevate temperature e pressione, D2O si è dimostrato molto efficiente per lo scambio H-D nei composti organici, evidenziando una differente reattività e selettività in dipendenza del pH.
La presenza di ola acqua deuterata induce solo parziale deuterazione di composti aromatici attivati; tale deuterazione è diretta verso sostituzioni elettrofiliche (es.orto-para deuterazione di aniline e fenoli)[66,67] e non risulta efficace nel promuovere lo scambio H-D in composti attivati debolmente come gli idrocarburi aromatici.
L'aggiunta di acidi[68,69] (es. DCl 2%) o di basi[70,71] (es. NaOD 2%) catalizza lo scambio equo di H-D negli idrocarburi e deattiva composti come l'acido benzoico, producendo il corrispondente composto perdeuterato.
Tuttavia, la procedura a temperatura elevata
dell'acido diluito (HTDA) richiede un contenitore di reazione resistente
all'acido[72] ed i prodotti alcalini di D2O richiedono temperature
di 380-
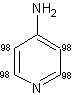
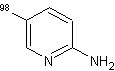
La reazione di scambio sui derivati della piridina procede molto bene al livello del nucleo della piridina in modo da dare i prodotti multi-deuterati desiderati sia in condizioni di soddisfacente efficienza del deuterio, sia in casi isolati.
In
particolare, è stato dimostrato da vari studi[73] che i derivati
della piridina quali l'amminopiridina e l'idrossipiridina hanno mostrato un notevole ed eccellente rendimento
di deuterazione a 160-
In generale, come mostra la (Tab.2.1), è stata osservata una più alta efficienza della deuterazione nelle posizioni adiacenti agli atomi di azoto sull'anello piridinico piuttosto che in altre posizioni.
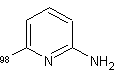
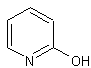
Dall'altra
parte, è stata osservata, presumibilmente a causa di impedimenti sterici,
un'inferiore incorporazione del deuterio al livello delle posizioni vicine al
sostituente (le posizioni orto), come ad esempio ![]() .
.
In generale, da studi fatti la reazione di scambio H-D procede in maniera efficiente anche in posizione orto rispetto al sostituente se la posizione è adiacente all'atomo dell'anello piridinico. E' evidente che l'atomo di azoto influenza profondamente la reazione di deuterazione usando il sistema Pd/C-H2-D2O.
|
Entry |
Temperatura (C°) |
D contenuto (%) |
Campo (%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tab. 2.1 Reazione di scambio H-D sui derivati della piridina in D2O catalizzato
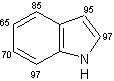
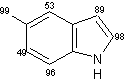
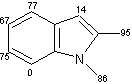
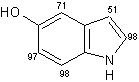
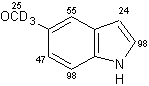
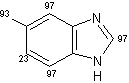
Un eccellente incorporazione del deuterio è stata osservata a livello dei sostituenti metilici come pure a livello delle posizioni adiacenti agli atomi di azoto sull' indolo, sull'azaindolo, benzimidazolo, ed anelli di chinolina, mentre l'efficienza della deuterazione nelle posizioni vicine dei gruppi metili si è rivelata generalmente bassa rispetto alla posizione adiacente al gruppo ossidrile. Inoltre, come mostra la (Tab 2.2), la posizione 7 dell'indolo e gli anelli benzimidazolo, che sono considerati essere in posizione orto rispetto ai gruppi amminici degli anelli benzenici sono stati deuterati in modo efficiente (entry 1-3, 6, 7, e 10). Da altra parte, nessuna incorporazione è stata trovata a livello della posizione di 7 quando l'1,2-dimetilindolo è stato utilizzato come un substrato (entry 5). I risultati riportati in precedenza dimostrano che questo metodo di deuterazione è fortemente influenzato da fattori sia sterici che elettronici.
|
Entry |
Temperatura (C°) |
D contenuto (%) |
Campo (%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tab.2.2 Reazione di scambio H-D sui derivati dell'indolo in D2O catalizzato
2.2.3 Reazione di scambio H-D dei derivati della Pirimidina ed immidazolo
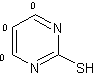
Quando la
2-mercaptopirimidina è stato utilizzato come substrato, non è stata osservata
nessuna reazione di scambio H-D e la dimerizzazione
procedeva come risultato della formazione di un legame disolfuro
(Tab.2.1 entry 1)
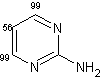
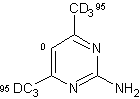
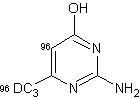
D'altra parte, quando il gruppo tiolo è stato sostituito con un gruppo amminico, la deuterazione, soprattutto a livello delle posizioni 4 e 6, è avvenuta con alte rese senza difficoltà. (entry 2).
Quando i due gruppi metilici sono stati introdotti nelle posizioni 4 e nella posizione 6 della 2-amminopirimidina non fu osservata alcuna incorporazione in posizione 5, mentre la posizione 5, che era adiacente al gruppo ossidrile, è stata quantitativamente deuterata quando la 2-ammino-4-idrossi-6-metilpirimidina è stata utilizzata come substrato (entry 3).
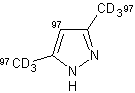
Inoltre, l'impiego del 3,5-dimetilpirazolo ha portato quasi ad un'incorporazione di deuterio quantitativamente importante in posizione 4 (entry 5).
Questi risultati suggeriscono che l'introduzione di un adeguato sostituente all'interno del substrato può permettere di stabilire una reazione di scambio H-D regioselettiva sfruttando l'ingombro sterico e/o gli effetti vicini del sostituente.
|
Entry |
Temperatura (C°) |
D contenuto (%) |
Campo (%) |
|
|
|
|
NA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tab.2.3 Reazione di scambio H-D sui derivati della Pirimidina ed imidazolo in D2O catalizzato
Le applicazioni di isotopi stabili classificati (SI) come composti per gli studi di farmacocinetica clinica e l'analisi di residui agrochimici per l'ambiente sono rapidamente aumentati negli ultimi anni.
Dal momento che le proprietà chimiche di composti classificati SI sono simili a quelle dei composti non classificati, i composti classificati come SI sono i più preziosi rivelatori per questi studi e analisi con GC-MS o LC-MS.
Nonostante l'utilità degli isotopi classificati (SI) come rivelatori, ci sono spesso problemi per ottenere il tracciante desiderato a causa della difficoltà nella sintesi dei composti SI-classificati.
Il riscaldamento di una varietà di composti
biologicamente attivi
composti nel sistema Pd/C-H2-D2O ha portato
all'efficiente
introduzione di atomi di deuterio. I risultati sono riassunti
nella (Tab 2.4)
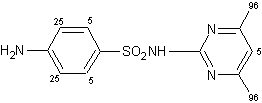
Le reazioni di scambio H-D a livello dei gruppi metilici nella sulfametazina e nell'acido nalidixico, entrambi agenti antibatterici, sono proseguite efficacemente ma l'efficienza della deuterazione preso del gruppo metilico dell'antipirina, un agente analgesico, è stata gravemente ridotta.
Invece, l'antipirina fu
deuterata in maniera regioneselettiva
sull'anello pirazolidinico (entry 4). Quando l'allopurinolo, un agente
antiurolitico, è stato utilizzato come un substrato, la reazione di scambio H-D
è avvenuta in modo efficace.
I farmaci deuterati hanno azioni spesso diverse dalle forme protonate in vivo.12. Alcuni farmaci deuterati mostrano diversi processi di trasporto.
Dal momento che molti farmaci deuterati risultano più resistenti ai cambiamenti metabolici grazie all'effetto dell'isotopo dovuto all'ingombro del deuterio, ci si può attendere che dai farmaci deuterio classificati si possano sviluppare dei farmaci con un nuovo dosaggio a rilascio prolungato in virtù dell'effetto dell'isotopo.
È stato dimostrato da studi recenti che il metodo della deuterazione potrebbe essere un metodo generale per la preparazione di nuovi farmaci a rilascio prolungato così come per materiale standard per gli studi del metabolismo e l'analisi dei residui di sostanze chimiche nell'ambiente.
|
Entry |
Temperatura (C°) |
D contenuto (%) |
Campo (%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tab. 2.4 Reazione di scambio H-D di composti biologicamente attivi in D2O catalizzato
Molte delle reazioni di scambio di idrogeno come scambio di H in composti etero atomici, oppure lo scambio di idrogeno nei chetoni ecc, sono catalizzate da acidi. Lo scambio di chetoni e di altri gruppi carbonilici è di importanza sufficiente, anche se la catalisi acida in scambio di idrogeno di idrogeno su gruppi ossidrilici è stato attentamente studiato, da Grunwald e suoi collaboratori, per misurare la velocità di cambio tra le molecole di metanolo in metanolo[75] e fenolo in metanolo[76].
Le
reazioni sono in ogni caso estremamente rapide, la velocità costante dei
trasferimenti di idrogeno acidi catalizzati tra molecole di metanolo deve essere
di ![]() a 80°C[75].
a 80°C[75].
Nel 1934, Ingol et.al.[77] ha riferito che il benzene scambia i suoi atomi idrogeno con deuterio in presenza di acido deuterosolfurico.
Ingol suppone che il meccanismo sia lo stesso di quello per le sostituzioni elettrofile aromatiche[77,78], mostrando che lo scambio di idrogeni nell'anisolo in posizione (orto e para) avviene più rapidamente[79].
Inoltre, si è osservato che con un acido sufficientemente forte il deuterio potrebbe anche essere introdotto in idrocarburi alifatici saturi, anche se con meno di selettività che in composti aromatici[80].
L'introduzione del deuterio nello scambio di reazione acido-catalizzata dipende molto dalla basicità del substrato.
Si è osservato che misure della basicità dei sistemi aromatici, dipendono dal seguente equilibrio:
![]()
Lo ione carbonio negli intermedi di reazioni di sostituzioni elettrofile aromatiche può essere scritto nel seguente modo:
![]()
questo ione viene caratterizzato da prodotti intermedi di reazioni acido catalizzata con scambio di deuterio che coinvolgono gli aromatici.
Un esempio non è altro che il mesitilene; infatti 1 mol di mesitilene, di trifluoruro di boro e fluoruro deuterato producono un complesso sigma avente la seguente struttura:
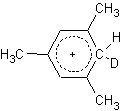
quindi possiamo dire che le regole che governano la reazione di scambio acido catalizzata del deuterio per certi versi è proprio uguale alla sostituzione aromatica.
La velocità della reazione dipende dall'acido forte[81] e, di conseguenza, dalle sostituzioni sull'anello aromatico.
Il metodo è sufficientemente utile, quando ci sono alte direttive influenzante nell'anello, come per esempio, nel p- nitrotoluene, che viene convertito nel seguente composto di deuterato:
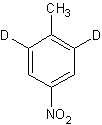
In considerazione della facilità di trasformazione del nitro gruppo ad un gruppo ammino ed ad altri sostituenti, questo metodo presenta un approccio molto conveniente per un gran numero di composti aromatici sostituiti.
Sia l'alogenazione base-catalizzata che la deuterazione di chetoni continuano con lo stesso step di reazione con la formazione dell'anione enolato (o enolo).
Ciò significa che l'orientamento e la velocità di reazione per le due reazioni sarebbero quasi identiche[82;83]. Di seguito viene riportato lo studio dell'alogenazione e la deuterazione base catalizzata,del 2- butanone[84;85].
Studi evidenti dimostrano che esistono due differenti alogenazioni base-catalizzate per il 2-butanone e, inaspettatamente, in nessuno di queste alogenazioni vi è lo stesso orientamento di quello della deuterazione[86].
Ciò conduce all'ipotesi che vi sono differenti meccanismi per alogenazione e la deuterazione.
Le alogenazioni hanno effettuato esperimenti in D20 dove l'acido ipobromoso è stato usato come agente alogenante e si è notato che la catalisi basica e l'alogenazione deuterata dei chetoni non sono rispettivamente reazioni competitive.
È stato suggerito, inoltre casi in cui, i meccanismi non enolici funzionassero nell'alogenazione; quindi è evidente che l'enolizazzione dei chetoni è stata studiata per favorire una serie di reazioni come la racemizzazione e la deuterazione, ma come già accennato in precedenza non per favorire le reazioni di alogenazione.
La deuterazione del 2 -butanone è stato studiato da warkentin e da Tee[87;88], essi hanno dimostrato che basi differenti hanno segnalato valori differenti per l'orientamento della deuterazione, come l'acetato di sodio, il p-nitrofenossido di sodio oppure il sodio deuterato[87;88].
Questo risultato è in disaccordo con le ipotesi sperimentali di Bothner e Sun in cui hanno usato il sodio acetato ed altre basi inorganiche[89] nella deuterazione come base sui loro composti deuterati.
in uno studio approfondito si è studiato la deuterazione base-catalizzata di un certo numero di metil chetoni tranne però per il 2-butanone, usando come tecnica spettroscopica l'NMR, dove le velocità calcolate e le costanti sono riassunti in (Tab. 2.5).
|
Chetoni-d |
CH3 |
CH2 |
CH |
KD |
|
Acetone |
|
|
|
|
|
2-butanone |
|
|
|
|
|
2-pentanone |
|
|
|
|
|
2-esanone |
|
|
|
|
|
2-eptanone |
|
|
|
|
|
3-metil-2butanone |
|
|
<10-2 |
<10-2 |
|
3-metil-2 pentanone |
|
|
<10-2 |
<10-2 |
|
4,4-dimetil-2-pentanone |
|
|
|
|
Tab. 2.5 Velocità delle costanti "KD" di deuterazione
Si è calcolato inoltre i valori di KD che rappresentano una misura della dell'orientamento della deuterazione. Questo valore di KD è definito come 3 deuterazione /1deuterazione KCH2/KCH3 oppure come KCH/KCH4.
Infatti dalla tabella 2.5 si può notare, che, esami fatti sulla velocità di deuterazione l'acetone risultato essere il chetone più reattivo, mentre la velocità dei chetoni non ramificati risulta essere circa la metà di quelli dell'acetone mentre i chetoni ramificati sono molto più bassi come nel caso del 4,4-dimetil-2-pentanone che è circa 20 volte inferiore all'acetone, che la deuterazione di un gruppo metilene o di un gruppo metino è molto più lenta di un gruppo metilico.
[59] A. LAI, M. CASU, G. SABA, F.P. CORONGIU, M.A. DESSI, Magn. Reson. Chem. 33, 163-l66 e riferimenti ivi inclusi (1995).
[60] M. DEIANA, F.P. CORONGIU, M.A. DESSI, P. SCANO, M. CASU, A. LAI, Magn. Reson. Chem. 39, 29-32 e riferimenti ivi inclusi (2001).
[61] STEACIEE,. CV. R. and co-workers. To be published.
For review, see for example: (a) Junk, T.; Catallo, W. J. Chem. Soc. Rev.
(b) Elander, N.; Jones, J. R.; Lu, S.-Y.; Stone-Elander, S. Chem. Soc. Rev.
For examples, see: (a) Campbell, R. E., Jr.; Lochow, C. F.; Vora, K. P.; Miller, R. G. J. Am. Chem. Soc.
(b) Baba, S. Radioisotopes
(c) Baldwin, J. E.; Adlington, R. M.; Ting, H.-H.; Arigoni, D.; Graf, P.; Martinoni, B. Tetrahedron
(d) Stevenson, D. E.; Akhtar, M.; Gani, D. Tetrahedron Lett.
(e) Hawthorne, S. B.; Miller, D. J.; Aulich, T. R. Fresenius Z. Anal. Chem.
(f) Furuta, T.; Takahashi, H.; Kasuya, Y. J. Am. Chem. Soc.
(g) Sellmann, D.; K appler, J.; Moll, M. J. Am. Chem. Soc.
(h)
(i) Gardner, K. H.; Kay, L. E. J. Am. Chem. Soc.
(j) Liu, K.; Williams, J.; Lee, H.; Fitzgerald, M. M.; Jensen, G. M.; Goodin, D. B.; McDermott, A. E. J. Am. Chem. Soc.
(k) Sack, I.; Balazs, Y. S.; Rahimipour, S.; Vega, S. J. Am. Chem. Soc.
(l) Takahashi, H.; Nakanishi, T.; Kami, K.; Arata, Y.; Shimada, I. Nat. Struct. Biol.
(m) Chou, M.-Y.; Mandal,A. B.; Leung, M.-K. J. Org. Chem.
(n)Durazo, A.; Abu-Omar, M. M. Chem. Commun.
Sajiki, H.; Hattori, K.; Aoki, F.; Yasunaga, K.; Hirota, K. Synlett
(a) Sajiki, H.; Aoki, F.; Esaki, H.; Maegawa, T.; Hirota, K. Org.Lett.
(b) Sajiki, H.; Esaki, H.; Aoki, F.;Maegawa, T.; Hirota, K. Synlett
(c) Esaki, H.; Aoki, F.; Maegawa, T.; Hirota, K.; Sajiki, H. Heterocycles
(d) Maegawa, T.; Akashi, A.; Esaki, H.; Aoki, F.; Sajiki, H.; Hirota, K. Synlett
(e) Sajiki, H.; Ito, N.; Esaki, H.; Maesawa, T.; Maegawa, T.; Hirota, K. Tetrahedron Lett.
(f) Ito, N.; Watahiki, T.; Maesawa, T.; Maegawa, T.; Sajiki, H. Adv. Synth. Catal.
[66]
[67] Bryson, T. A.;
[68] Werstiuk, N. H.; Kadai, T. Can. J. Chem. 1974, 52, 2169-2171.
[69] Werstiuk, N. H.;
[70]
[71] Junk, T.; Cavallo, W. J. Tetrahedron Lett. 1996, 37, 3445-3448.
[72] Buncel, E.; Jones, J. R. Isotopes in the Physical and Biomedical
Sciences. Volume 1.
Labelled Compounds (Part A); Elsevier:
[73] Campbell, R. E., Jr.; Lochow, C. F.; Vora, K. P.; Miller, R. G. J. Am. Chem. Soc.
[74] Allen I.Fernstein and Ellis K. Fields; A Novel Intramolecular Free-Radical Cyclization in the Vapor-Phase Arylation of Methyl Benzoate; J. Org. Chem., Vol. 37, No. 1, 1972.
E.K. Fields
and
[75] E.Grunwald, C. F. Jumper, and S. Meiboom, J.Am.Chem. Soc., 84, 4664 (1962). Kinetic of proton transfer in methanol and the mechanism of the abnormal conductance of the hydrogen ion.
[76]
E.Grunwald, C. F. Jumper, and
[77] C. K.Ingol, C. G. Raisin and C. L. Wilson, Nature, 134, 847 (1934). Direct introduction of deuterium into benzene.
[78] C. K.Ingol, C. G. Raisin and C. L. Wilson, Chem. Soc., 915, (1936). Structure of benzene. Part II. Direct introduction of deuterium into benzene and the physical properties of exadeuterobenzene.
[79] C.
K.Ingol, C. G. Raisin and C. L. Wilson, Chem. Soc., 1637, (1936). Direct
introduction of deuterium into the aromatic nucleus.
[80] C.
K.Ingol, C. G. Raisin and C. L. Wilson, Chem. Soc., 1643, (1936). Direct
introduction of deuterium into aliphatic system.
[81] A.F. Thomas and B.Willhalm, Tet. Letters, 5129 (1967). Mass spectrometry and organic analysis. XI. The mass spectrum of B-ionone.
C. K.
Ingold, "Structure and Mechanism in Organic Chemistry,"
D. J. Cram. "Fundamentals of
Carbanion Chemistry," Academic Preas Inc.,
[84] C. Rappe, Acta Chem. Scond.,
[85] C. Rappe, ibid., in press.
[86] C. Rappe, ibid.,
[87] J. Warkentin and S. Tee, Chem. Cornnun., 190 (1966).
[88] J. Warkentin and S. Tee, J . Am. Chem. Soc., 88,
|
Privacy
|
© ePerTutti.com : tutti i diritti riservati
:::::
Condizioni Generali - Invia - Contatta